聯 系 人 :秦經理
座 機(jī) :0515-84130555
手 機(jī):17705109088
郵(yóu) 箱:info@gtaipeptide.com
地 址:江蘇(sū)省鹽城市濱(bīn)海縣新安大道799号
網(wǎng) 址:www.36467.cn
郵(yóu) 編(biān):224500
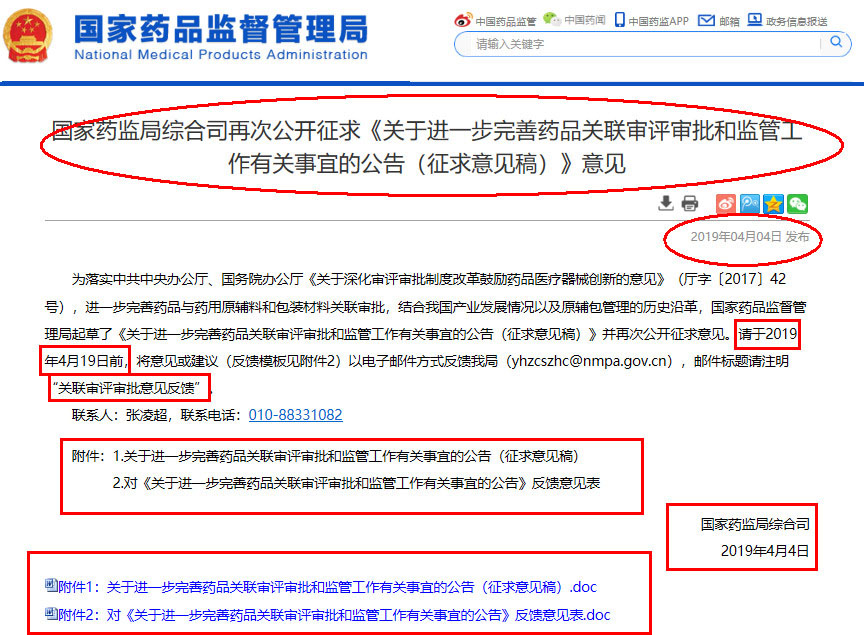
國家藥監局綜合司再次公開征求《關(guān)於(yú)進一步完善藥品關(guān)聯審評審批和監管工作有關(guān)事宜的公告(征求意見稿)》意見 |
爲落實中共中央辦公廳、國務院辦公廳《關於深化審評審批制度改革鼓勵藥品醫療器械創新的意見》(廳字〔2017〕42号),進一步完善藥品與藥用原輔料和包裝材料關聯審批,結合我國産業發展情況以及原輔包管理的曆史沿革,國家藥品監督管理局起草瞭《關於進一步完善藥品關聯審評審批和監管工作有關事宜的公告(征求意見稿)》並再次公開征求意見。請於2019年4月19日前,将意見或建議(反饋模闆見附件2)以電子郵件方式反饋我局(yhzcszhc@nmpa.gov.cn),郵件标題請注明“關聯審評審批意見反饋”。 附件:1.關於進一步完善藥品關聯審評審批和監管工作有關事宜的公告(征求意見稿) 國家藥監局綜合司
|
附件1
關於進一步完善藥品關聯審評審批和監管工作
有關事宜的公告
(征求意見稿)
爲落實中共中央辦公廳、國務院辦公廳《關於深化審評審批制度改革鼓勵藥品醫療器械創新的意見》(廳字〔2017〕42号),原食品藥品監管總局發布瞭《關於調整原料藥、藥用輔料和藥包材審評審批事項的公告》(2017年第146号),爲進一步明確原料藥、藥用輔料、藥包材(以下簡稱原輔包)與藥品關聯審評審批和監管有關事宜,保障原輔包供應及合規使用,現就有關事項公告如下:
一、總體要求
(一)原輔包與藥品的關聯審評審批可由原輔包登記人在登記平台上登記,藥品制劑申請人提交注冊申請時與平台登記資料進行關聯;也可在藥品制劑注冊申請時,由藥品制劑申請人一並提供原輔包研究資料;原則上對原輔包不再單獨審評審批,但仿制國内已上市的原料藥除外。
(二)原輔包在登記平台登記,方便藥品制劑申請人進行選擇,爲藥品制劑産品研發提供更多支持,同時也避免原輔包所有權人在審評過程中反複提供研究資料,減輕企業負擔。
(三)原輔包登記人應履行登記人相關義務,維護登記平台原輔包信息,並對其登記資料的真實性負責。國内原輔包生産企業一般應作爲原輔包登記人對所持有的産品自行登記。境外原輔包供應商可以直接登記,也可委托1家中國境内的法人機構作爲全權代理人進行登記,登記資料應當爲中文。原輔包登記委托人和代理機構共同對登記資料的真實性負責。
(四)藥品制劑申請人申報藥品注冊申請時,使用登記平台的原輔包資料的,隻需提供原輔包的登記号和原輔包登記人的使用授權書。若使用的原輔包未在登記平台登記,則需要與制劑注冊申請一並提供相應原輔包全套研究資料。
(五)藥品上市許可持有人對藥品質量承擔主體責任,對選用的原輔包質量及合法性負責,根據注冊管理和上市後生産管理的要求,對原輔包生産企業進行管理,保證藥品質量。
(六)監管部門對原輔包登記人提交的技術資料負有保密責任,對登記平台的技術信息保密。平台隻公開登記品種的基本信息(包括企業基本信息)和登記狀态标識。
二、産品登記管理
(七)原輔包按照登記資料技術要求在平台登記,完成後獲得登記号。其中,原料藥登記按照原食品藥品監管總局《關於發布化學藥品新注冊分類申報資料要求(試行)的通告》(2016年第80号)要求執行,並應在登記前取得《藥品生産許可證》;藥用輔料和藥包材登記按照本公告附件1、附件2執行。部分資料不能提供的,在說明理由後予以登記。原輔包登記操作具體要求在藥審中心官方網站公布,並及時更新。
(八)藥品制劑注冊申請與已登記原輔包進行關聯的,藥品制劑獲得批準時,即表明該關聯的原輔包通過技術審評,在登記平台标識爲“A”;未通過技術審評或尚未與制劑注冊進行關聯的标識爲“I”。
關聯審評審批政策實施前曾經取得批準證明文件並持續在制劑中使用的的原輔包,由國家藥品監督管理局将批準信息轉入登記平台並給予登記号,登記狀态标識爲“A”,可繼續在原藥品中使用,國家公布禁止使用或者淘汰的原輔包除外。
(九)仿制國内已上市的原料藥,登記時可以不與藥品制劑關聯,即單獨登記,獨立審評,經審評通過後,登記狀态标識爲“A”,未通過審評的标識爲“I”。時限按照現行《藥品注冊管理辦法》執行。
(十)完成登記的原輔包的基本信息在登記平台公示,包括登記狀态标識(A或I)和産品基本信息。産品基本信息包括登記号、品種名稱、企業名稱(代理機構名稱)、企業地址、産品來源、規格、更新日期和其他必要的信息。
(十一)已在食品、藥品中長期使用且安全性得到證明的藥用輔料可不進行登記(名單詳見附件3),由藥品制劑申請人在制劑申報資料中列明産品清單和基本信息。但藥審中心在藥品制劑技術審評過程中認爲有必要的,可要求藥品制劑申請人提供相應技術資料。藥審中心将根據工作需要适時對該類産品名單進行更新。
(十二)藥用輔料、藥包材已取消行政許可,平台登記不收取費用。原料藥仍爲行政許可,平台登記和技術審評費用按原标準執行。
三、原輔包登記信息的使用和管理
(十三)審評藥品制劑注冊申請時,原輔包研究資料不能滿足審評需要的,審評機構應要求藥品制劑申請人或原輔包登記人進行補正。補正資料可以由藥品制劑申請人直接遞交藥審中心,也可由原輔包登記人通過平台補充完善登記信息。具體工作程序由藥品審評部門制定公布。
(十四)原料藥标識爲“A”的,表示原料藥通過審評審批,但不再發給藥品批準文号,原料藥登記人可以自行在登記平台打印《藥品注冊批件》《進口藥品注冊證》或《醫藥産品注冊證》,用於辦理GMP認證、藥品進口通關等。該批準證明文件有效期與登記平台狀态标識相關聯。
與制劑申報資料一並提交研究資料的原料藥,監管部門在制劑批準證明文件中标注原料藥相關信息,可用於辦理原料藥GMP認證、藥品進口通關等。
(十五)原料藥生産企業申請GMP認證程序及要求按照現行法律法規有關規定執行,取得藥品GMP證書後應在登記平台更新登記信息。
(十六)審評通過的原輔包登記信息可以進行補充完善和變更。原輔包登記信息應當包含場地管理文件信息,具體文件格式另行發布。原料藥發生技術變更的,要提交變更申請。原料藥的非技術類變更、藥用輔料和藥包材的重大變更和中等變更應及時在登記平台更新信息。原輔包的微小變更可以在年度報告中一並提交。具體工作程序由藥品審評部門制定公布。
(十七)原輔包的任何變更均需及時告知相關藥品制劑申請人(藥品上市許可持有人),藥品制劑申請人(藥品上市許可持有人)應就變更對制劑質量的影響情況進行評估或研究。
(十八)已上市制劑變更原輔包供應商的,應按照《已上市化學藥品變更研究技術指導原則(一)》《已上市化學藥品生産工藝變更研究技術指導原則》《已上市中藥變更研究技術指導原則(一)》及生物制品上市後變更研究相關指導原則等要求開展研究。其中拟使用的原輔包登記狀态标識爲“I”的,應報藥審中心進行關聯審評審批;拟使用的原輔包登記狀态标識爲“A”的,但相應制劑爲《上市藥品目錄集》收載的藥品和進口藥品,需要提交補充申請報藥審中心審評,其他制劑變更原輔包标識爲“A”的供應商報省局備案。補充申請或備案申請均應提交相關研究資料。
(十九)境外原輔包供應商更換代理機構的,提交相關證明性資料後予以變更(提供境外廠商委托新的代理機構申報的委托書、公證文書及其中文譯本、新的代理機構營業執照複印件,境外廠商解除原代理機構委托關系的文書、公證文書及其中文譯本)。
四、監督管理
(二十)各省(區、市)藥品監督管理局對獲得關聯審評審批的原料藥(登記狀态标識爲“A”的)按照藥品進行上市後管理,生産企業需取得《藥品生産許可證》,按照藥品生産質量管理規範要求開展認證檢查。
(二十一)各省(區、市)藥品監督管理局應加強對本行政區域内制劑企業的監督檢查,督促制劑企業履行藥用輔料和藥包材的供應商審計責任。藥用輔料和藥包材生産企業具有《藥品生産許可證》的,各省(區、市)藥品監督管理局繼續按原管理要求管理,許可證到期後不再換發新證,對其生産場地按本公告要求進行管理。
(二十二)各省(區、市)藥品監督管理局可根據登記信息對藥品上市許可持有人所使用的藥用輔料和藥包材供應商開展有因延伸檢查。發現藥用輔料和藥包材生産存在質量問題的,應要求制劑企業不得使用相關産品,並對已上市産品開展評估和處置。延伸檢查可由制劑企業所在地省局組織開展,也可由其聯合藥用輔料和藥包材生産企業所在地省局開展。
(二十三)藥用輔料生産現場檢查按《藥用輔料生産質量管理規範》開展檢查,藥包材生産現場檢查按《直接接觸藥品的包裝材料和容器管理辦法》中所附《藥包材生産現場考核通則》開展檢查。
五、其他
(二十四)本公告自xx年xx月xx日起實施。原發布的原輔包相關文件與本公告不一緻的,以本公告爲準。原國家食品藥品監督管理總局發布的《關於發布藥包材藥用輔料申報資料要求(試行)的通告》(2016年第155号)同時廢止。
特此公告。
附件:1.藥用輔料研究資料要求(試行)
2.藥包材研究資料要求(試行)
3.可免登記的産品目錄
4.藥用原輔料、藥包材年度報告基本要求
國家藥監局
附件1
藥用輔料研究資料要求(試行)
品種名稱:XXXXX
登 記 人:XXXXX
輔料分類:
境内外上市藥品中未有使用曆史的,包括
○1.1 新的分子結構的輔料以及不屬於第1.2、1.3的輔料;
○1.2 由已有使用曆史的輔料經簡單化學結構改變(如鹽基,水合物等);
○1.3 兩者及兩者以上已有使用曆史的輔料經共處理得到的輔料;
○1.4 已有使用曆史但改變給藥途徑的輔料。
境内外上市藥品中已有使用曆史的,且
○2.1 中國藥典/USP/EP/BP/JP均未收載的輔料;
○2.2 USP/EP/BP/JP之一已收載,但未在境内上市藥品中使用的輔料;
○2.3 USP/EP/BP/JP之一已收載,中國藥典未收載的輔料;
○2.4 中國藥典已收載的輔料。
在食品或化妝品中已有使用曆史的,且
○3.1 具有食品安全國家标準的用於口服制劑的輔料;
○3.2 具有化妝品國家或行業标準的用於外用制劑的輔料。
○其他
拟用制劑給藥途徑:○注射 ○吸入 ○眼用 ○局部及舌下 ○透皮 ○口服 ○其他
來源:○動物或人 ○礦物 ○植物 ○化學合成 ○微生物發酵或生物工程 ○其他
登記人名稱: | 蓋章 | |
法定代表人: | 簽名 |
一、登記資料項目
1 登記人基本信息
1.1 登記人名稱、地址、生産地址
1.2 證明性文件
1.3 研究資料保存地址
2 輔料基本信息
2.1 名稱
2.2 結構與組成
2.3 理化性質及基本特性
2.4 境内外批準上市及使用信息
2.5 國内外藥典收載情況
3 生産信息
3.1 生産工藝和過程控制
3.2 物料控制
3.3 關鍵步驟和中間體的控制
3.4 工藝驗證和評價
3.5 生産工藝的開發
4 特性鑒定
4.1 結構和理化性質研究
4.2 雜質研究
4.3 功能特性
5 質量控制
5.1 質量标準
5.2 分析方法的驗證
5.3 質量标準制定依據
6 批檢驗報告
7 穩定性研究
7.1 穩定性總結
7.2 穩定性數據
7.3 輔料的包裝
8 藥理毒理研究
二、登記資料正文及撰寫要求
1 登記人基本信息
1.1 登記人名稱、注冊地址、生産地址
提供登記人的名稱、注冊地址、生産廠、生産地址。
生産地址應精確至生産車間、生産線。
1.2 證明性文件
境内藥用輔料登記人需提交以下證明文件:
(1)登記人營業執照複印件。對登記人委托第三方進行生産的,應同時提交委托書等相關文件、生産者相關信息及營業執照複印件。
(2)對於申請藥用明膠空心膠囊、膠囊用明膠和藥用明膠的境内登記人,需另提供:①申請藥用空心膠囊的,應提供明膠的合法來源證明文件,包括藥用明膠的批準證明文件、标準、檢驗報告、藥用明膠生産企業的營業執照、《藥品生産許可證》、銷售發票、供貨協議等的複印件;②申請膠囊用明膠、藥用明膠的,應提供明膠制備原料的來源、種類、标準等相關資料和證明。
境外藥用輔料登記人應授權中國代表機構提交以下證明文件:
(1)登記人合法生産資格證明文件、公證文件及其中文譯文。對登記人委托第三方進行生産的,應同時提交委托書等相關文件及生産者相關信息及證明文件(如有)。
(2)登記人委托中國境内代理機構注冊的授權文書、公證文件及其中文譯文。中國境内代理機構的營業執照或者登記人常駐中國境内辦事機構的《外國企業常駐中國代表機構登記證》。
(3)登記藥用空心膠囊、膠囊用明膠、藥用明膠等牛源性藥用輔料進口的,須提供制備膠囊的主要原材料——明膠的制備原料的來源、種類等相關資料和證明,並提供制備原料來源於沒有發生瘋牛病疫情國家的政府證明文件。
境外藥用輔料建議提供人源或動物源性輔料的相關證明文件。
1.3 研究資料保存地址
提供藥用輔料研究資料的保存地址,應精確至門牌号。如研究資料有多個保存地址的,均需提交。
2 輔料基本信息
2.1 名稱
提供輔料的中文通用名(如适用,以中國藥典名爲準)、英文名、漢語拼音、化學名、曾用名、化學文摘(CAS)号等。如有UNII号及其他名稱(包括國内外藥典收載的名稱)建議一並提供。
預混輔料[注1]和共處理輔料[注2]應明確所使用的單一輔料並進行定性和定量的描述,可提交典型配方用於說明,實際應用的具體配方應根據使用情況作爲附件包括在登記資料中或在藥品注冊時進行提供。
注1:預混輔料(pre-mixed excipient)是指兩種或兩種以上輔料通過低至中等剪切力進行混合,這是一種簡單的物理混合物。各組分混合後仍保持爲獨立的化學實體,各成分的化學特性並未變化。預混輔料可以是固态或液态,單純的物理混合時間較短。
注2:共處理輔料(co-processed excipient)是兩種或兩種以上輔料的結合物,該結合物的物理特性發生瞭改變但化學特性無明顯變化。這種物理特性的改變無法通過單純的物理混合而獲得,在某些情況下,有可能以成鹽形式存在。
2.2 結構與組成
提供輔料的結構與組成信息,如結構式、分子式、分子量,高分子藥用輔料應明確型号、分子量範圍、聚合度、取代度等。有立體結構和多晶型現象應特别說明。
預混輔料和共處理輔料應提交每一組分的結構信息。
2.3 理化性質及基本特性
提供輔料已知的物理和化學性質,如:性狀(如外觀,顔色,物理狀态)、熔點或沸點、比旋度、溶解性、溶液pH、粒度、密度(堆密度、振實密度等)以及功能相關性指标等。
預混輔料應提交産品性狀等基本特性信息。
2.4 境内外批準登記等相關信息及用途
2.4.1 境内曆史批準信息
提供境内曆史批準的相關信息(如有)。
2.4.2 其他國家的相關信息
提供拟登記産品在境外作爲藥用輔料的相關信息(如适用)。
2.4.3 用途信息
提供該輔料的給藥途徑信息以及最大每日參考劑量及參考依據。使用該輔料的藥品已在境内外獲準上市的,提供相關藥品的劑型、給藥途徑等;尚未有使用該輔料的藥品獲準上市的,應提供該藥用輔料的預期給藥途徑以及正在使用該輔料進行注冊的藥品信息。如有生産商已知的不建議的給藥途徑或限定的使用劑量,也應予以明確並提供相關參考說明。以上信息應盡可能提供。
2.5 國内外藥典收載情況
提供該藥用輔料被國内外藥典及我國國家标準收載的信息。
3 生産信息
3.1 生産工藝和過程控制
(1)工藝綜述:按工藝步驟提供工藝流程圖,並進行生産工藝綜述。
(2)工藝詳述:按工藝流程标明工藝參數和所用溶劑等。如爲化學合成的藥用輔料,還應提供反應條件(如溫度、壓力、時間、催化劑等)及其化學反應式,其中應包括起始原料、中間體、所用反應試劑的分子式、分子量、化學結構式。
以商業批爲代表,列明主要工藝步驟、各反應物料的投料量及各步收率範圍,明確關鍵生産步驟、關鍵工藝參數以及中間體的質控指标。
對於人或動物來源的輔料,該輔料的生産工藝中應有明確的病毒滅活與清除的工藝步驟,並須對其進行驗證。
(3)說明商業生産的分批原則、批量範圍和依據。
(4)設備:提供主要和特殊的生産設備。
生産設備資料可以按照下述表格形式提交:
生産設備一覽表
序号 | 設備名稱 | 型号 | 用途 | 生産商 | 生産範圍 |
1 | |||||
2 | |||||
… |
3.2 物料控制
3.2.1 關鍵物料控制信息
對關鍵物料的控制按下表提供信息。
關鍵物料控制信息
物料名稱 | 來源注 | 質量标準 | 使用步驟 | 生産商 | 規格 |
注:如動物來源、植物來源、化學合成等。
3.2.2 物料控制信息詳述
按照工藝流程圖中的工序,以表格的形式列明生産中用到的所有物料(如起始物料、反應試劑、溶劑、催化劑等),並說明所使用的步驟,示例如下。
物料控制信息
物料名稱 | 來源注 | 質量标準 | 使用步驟 | 生産商 | 規格 |
注:如動物來源、植物來源、化學合成等。
提供以上物料的來源、明確引用标準,或提供内控标準(包括項目、檢測方法和限度),必要時提供方法學驗證資料。
3.3 關鍵步驟和中間體的控制
列出關鍵步驟(如:終産品的精制、純化工藝步驟,人或動物來源輔料的病毒滅活/去除步驟)。适用時,提供關鍵過程控制及參數,提供具體的研究資料(包括研究方法、研究結果和研究結論),支持關鍵步驟確定的合理性以及工藝參數控制範圍的合理性。存在分離的中間體時,應列出其質量控制标準,包括項目、方法和限度,並提供必要的方法學驗證資料。
3.4 工藝驗證和評價
3.4.1 工藝穩定性評估
提供輔料工藝穩定的相關評估資料,如5批以上的産品質量回顧性報告等。
3.4.2 工藝驗證
提供工藝驗證方案、驗證報告等資料,必要時提供批生産記錄樣稿。
3.5 生産工藝的開發
提供工藝路線的選擇依據(包括文獻依據和/或理論依據)。
提供詳細的研究資料(包括研究方法、研究結果和研究結論)以說明關鍵步驟確定的合理性以及工藝參數控制範圍的合理性。
詳細說明在工藝開發過程中生産工藝的主要變化(包括工藝路線、工藝參數、批量以及設備等的變化)及相關的支持性驗證研究資料。提供工藝研究數據彙總表,示例如下:
工藝研究數據彙總表
批号 | 試制日期 | 試制地點 | 試制目的/樣品用途注1 | 批量 | 收率 | 工藝注2 | 樣品質量 | ||
含量 | 功能性指标 | 性狀等 | |||||||
注1:說明生産該批次的目的和樣品用途,例如工藝驗證/穩定性研究。
注2:說明表中所列批次的生産工藝是否與3.1項下工藝一緻,如不一緻,應明確不同點。
4 特性鑒定
4.1 結構和理化性質研究
4.1.1 結構確證研究
(1)結構確證信息
提供可用於對藥用輔料的結構進行確證或表征的相關信息。
(2)結構確證研究
應結合制備工藝路線以及各種結構確證手段對産品的結構進行解析,如可能含有立體結構、結晶水/結晶溶劑或者多晶型問題要詳細說明,對於高分子藥用輔料,還需關注分子量及分子量分布、聚合度、取代度、紅外光譜等結構確證信息。提供結構確證用樣品的精制方法、純度、批号;提供具體的研究數據和圖譜並進行解析。
爲瞭確保生物制品來源的藥用輔料質量的一緻性,需要建立标準品/對照品或将輔料與其天然類似物進行比較。對於生物制品類輔料具體見ICH關於生物技術/生物産品的指南。
對來源於化學合成體或來源於動/植物的預混輔料,需要用不同的方法描述其特性,並進行定量和定性的描述,包括所有特殊信息。
4.1.2理化性質
提供輔料理化性質研究資料,如:性狀(如外觀,顔色,物理狀态)、熔點或沸點、比旋度、溶解性、吸濕性、溶液pH、分配系數、解離常數、将用於制劑生産的物理形态(如多晶型、溶劑化物或水合物)、粒度、來源等。
4.2 雜質研究
4.2.1雜質信息
結合輔料生産工藝,描述雜質情況。
4.2.2 雜質研究
應根據藥用輔料的分子特性、來源、制備工藝等進行雜質研究,如對於高分子輔料,應重點研究殘留單體、催化劑以及生産工藝帶來的雜質。評估雜質對藥用輔料安全性、功能性等的影響,並進行相應的控制。
4.3 功能特性
4.3.1 功能特性信息
結合輔料在制劑中的用途及給藥途徑,提供輔料有關功能性指标信息(如适用)。
4.3.2 功能特性研究
結合輔料在制劑中的用途及給藥途徑,詳細說明該藥用輔料的主要功能特性並提供相應的研究資料。
如:粘合劑可提供表面張力、粒度及粒度分布、溶解性、粘度、比表面積、堆積度等适用的特性指标。
5 質量控制
5.1 質量标準
提供藥用輔料的質量标準。質量标準應當符合《中華人民共和國藥典》現行版的通用技術要求和格式,並使用其術語和計量單位。
5.2 分析方法的驗證
提供質量标準中有關項目的方法學驗證資料。對於現行版中國藥典/美國藥典/歐洲藥典/英國藥典/日本藥典已收載的品種,如採用藥典标準方法,可視情況開展方法學確認。
5.3 質量标準制定依據
說明各項目設定的考慮,總結分析各檢查方法選擇以及限度確定的依據。質量标準起草說明應當包括标準中控制項目的選定、方法選擇、檢查及純度和限度範圍等的制定依據。
6 批檢驗報告
提供不少於三批生産樣品的檢驗報告。如果有委托外單位檢驗的項目需說明。委托檢驗的受托方需具備相關資質。
7 穩定性研究
穩定性研究的試驗資料及文獻資料。包括採用直接接觸藥用輔料的包裝材料和容器共同進行的穩定性試驗。如适用,描述針對所選用包材進行的相容性和支持性研究。
7.1 穩定性總結
總結所進行的穩定性研究的樣品情況、考察條件、考察指标和考察結果,對各項指标變化趨勢進行分析,並提出貯存條件和有效期。
7.2 穩定性數據
以表格形式提供穩定性研究的具體結果,並将穩定性研究中的相關圖譜作爲附件。
7.3 輔料的包裝
說明輔料的包裝及選擇依據,提供包裝标簽樣稿。
8 藥理毒理研究
一般需提供的藥理毒理研究資料或文獻資料包括:
(1)藥理毒理研究資料綜述。
(2)對拟應用藥物的藥效學影響試驗資料或文獻資料。
(3)非臨床藥代動力學試驗資料或文獻資料。
(4)安全藥理學的試驗資料或文獻資料。
(5)單次給藥毒理性的試驗資料或文獻資料。
(6)重複給藥毒理性的試驗資料或文獻資料。
(7)過敏性(局部、全身和光敏毒性)、溶血性和局部(血管、皮膚、粘膜、肌肉等)刺激性等主要與局部、全身給藥相關的特殊安全性試驗研究或文獻資料。
(8)遺傳毒性試驗資料或文獻資料。
(9)生殖毒性試驗資料或文獻資料。
(10)緻癌試驗資料或文獻資料。
(11)其他安全性試驗資料或文獻資料。
根據藥用輔料的上市狀态、應用情況、風險程度等確定需提交的研究資料和/或文獻資料,如不需要某項研究資料時,應在相應的研究項目下予以說明。藥用輔料的藥理毒理研究可單獨進行也可通過合理設計與關聯制劑的藥理毒理研究合並進行。
三、登記資料說明
1 基於輔料(在制劑中)的使用曆史及藥典收載情況,附表1列出瞭不同類别輔料所需提供的資料文件。
2 登記資料應列出全部資料項目,對按附表1規定無需提供的資料,應在該項資料項目下進行說明。
3 對於之前按注冊程序已獲批準證明文件的藥用輔料,可按附表1第2.4類資料要求提供資料。審評過程中可根據需要補充資料。
4 輔料已有使用曆史的定義:該輔料已在境内外批準制劑中使用且給藥途徑相同。
5 境外批準制劑的範圍:僅限在美國、歐盟、日本批準上市的制劑。
6 對於分類未涵蓋的藥用輔料,請選擇“其它”,其登記資料的要求根據使用曆史和藥典收載情況提交相關的登記資料。
7 境内外上市藥品中已有使用曆史的,對於中國藥典已收載,USP/EP/BP/JP均未收載的藥用輔料參照2.1類提交登記資料。
8 對於同一輔料同時屬於不同分類的情況,應按照風險等級高的分類進行登記提交相關技術資料。
附表1
藥用輔料登記資料表
資料項目 | 内容 | 1.1* | 1.2* | 1.3* | 1.4* | 2.1* | 2.2* | 2.3* | 2.4* | 3.1* | 3.2* |
1 | 登記人基本信息 | + | + | + | + | + | + | + | + | + | + |
2 | 輔料基本信息 | + | + | + | + | + | + | + | + | + | + |
3 | 3.1(1)工藝綜述 | + | + | + | + | + | + | + | + | + | + |
3.1(2)工藝詳述 | + | ± | ± | ± | ± | ± | - | - | ± | ± | |
3.1(3)說明商業生産的分批原則、批量範圍和依據 | + | + | + | + | + | + | + | + | + | + | |
3.1(4)設備 | + | + | + | + | + | + | + | + | + | + | |
3.2.1 關鍵物料控制信息 | - | - | - | + | - | - | + | + | + | + | |
3.2.2 物料控制信息詳述 | + | + | + | - | + | + | - | - | - | - | |
3.3 關鍵步驟和中間體的控制 | + | + | + | + | + | + | - | - | + | + | |
3.4.1工藝穩定性評估 | - | - | - | + | + | + | + | + | + | + | |
3.4.2 工藝驗證 | + | + | + | - | - | - | - | - | - | - | |
3.5 生産工藝的開發 | + | ± | ± | - | ± | ± | - | - | - | - | |
4 | 4.1.1(1) 結構確證信息 | + | + | + | + | + | + | ± | - | + | + |
4.1.1(2)結構確證研究 | + | ± | + | - | - | - | - | - | - | - | |
4.1.2 理化性質 | + | ± | ± | ± | ± | ± | - | - | ± | ± | |
4.2.1 雜質信息 | + | + | + | + | + | + | + | + | + | + | |
4.2.2 雜質研究 | + | ± | ± | ± | ± | ± | - | - | ± | ± | |
4.3.1 功能特性信息 | + | + | + | + | + | + | + | + | + | + | |
4.3.2 功能特性研究 | + | + | + | ± | ± | ± | - | - | ± | ± | |
5 | 5.1 質量标準 | + | + | + | + | + | + | + | + | + | + |
5.2 分析方法的驗證 | + | + | + | ± | + | + | - | - | ± | ± | |
5.3 質量标準制定依據 | + | + | + | + | + | + | + | + | + | + | |
6 | 批檢驗報告 | + | + | + | + | + | + | + | + | + | + |
7 | 7.1 穩定性總結 | + | + | + | + | + | + | + | + | + | + |
7.2 穩定性數據 | + | + | + | + | + | + | + | + | + | + | |
7.3 輔料的包裝 | + | + | + | + | + | + | + | + | + | + | |
8 | 藥理毒理研究 | + | + | + | + | + | ± | ± | ± | ± | ± |
注:+ 需提供相關資料的項目
- 無需提供相關資料的項目
± 根據需要提供相關資料的項目
備注:*
境内外上市藥品中未有使用曆史的,包括
1.1 新的分子結構的輔料以及不屬於第1.2、1.3的輔料;
1.2 由已有使用曆史的輔料經簡單化學結構改變(如鹽基,水合物等);
1.3 兩者及兩者以上已有使用曆史的輔料經共處理得到的輔料;
1.4 已有使用曆史但改變給藥途徑的輔料。
境内外上市藥品中已有使用曆史的,且
2.1 中國藥典/USP/EP/BP/JP均未收載的輔料;
2.2 USP/EP/BP/JP之一已收載,但未在境内上市藥品中使用的輔料;
2.3 USP/EP/BP/JP之一已收載,中國藥典未收載的輔料;
2.4 中國藥典已收載的輔料。
在食品或化妝品中已有使用曆史的,且
3.1 具有食品安全國家标準的用於口服制劑的輔料;
3.2 具有化妝品國家或行業标準的用於外用制劑的輔料。
注:
(1)高風險藥用輔料一般包括:動物源或人源的藥用輔料;用於注射劑、眼用制劑、吸入制劑等的藥用輔料。對於高風險輔料的登記資料要求,可根據輔料在特定制劑中的應用以及相應的技術要求,按需提供,或在審評過程中根據特定制劑及輔料在制劑中的應用情況根據需要補充資料。
(2)對於已有使用曆史的輔料,若該輔料超出相應給藥途徑的曆史最大使用量,應提供相關安全性數據等資料。
(3)對預混輔料,應根據其在制劑中的應用及配方組成中各輔料成分情況,選擇合适的資料要求進行登記。
(4)國家食品藥品監督管理總局134号公告及其解讀中已規定不納入關聯審評審批的藥用輔料,仍不納入藥用輔料登記的範圍。
(5)以上登記資料分類要求作爲登記人資料準備的指導,藥品審評中心可根據制劑的技術審評需要提出資料補充要求。
(6)根據輔料分類不同,登記資料3.2.1與3.2.2,3.4.1與3.4.2,4.1.1(1)與(2)中提供一組研究資料即可。
附件2
藥包材研究資料要求(試行)
品種名稱:XXXXX
登 記 人:XXXXX
使用情況分類:
○1未在境内外上市藥品中使用過的藥包材(如新材料、新結構);
○2已在境内外上市藥品中使用過,但改變藥品給藥途徑且風險提高的藥包材;
○3未在境内外上市藥品中使用過,但是可證明在食品包裝中使用過的與食品直接接觸的藥包材(僅限用於口服制劑);
○4已在相同給藥途徑的上市藥品中使用過的藥包材
○4.1無注冊證的藥包材
○4.2有注冊證的藥包材
○5其他
登記人名稱: | 蓋章 | |
法定代表人: | 簽名 |
一、登記資料項目
1 登記人基本信息
1.1 名稱、地址、生産廠、生産地址
1.2 證明性文件
1.3 研究資料保存地址
2 藥包材基本信息
2.1 藥包材名稱
2.2 包裝系統/組件
2.3 配方
2.4 基本特性
2.5 境内外批準上市及使用信息
2.6 國家标準以及國内外藥典收載情況
3 生産信息
3.1 生産工藝和過程控制
3.2 物料控制
3.3 關鍵步驟和半成品/中間體的控制
3.4 工藝驗證和評價
4 質量控制
4.1 質量标準
4.2 分析方法的驗證
4.3 質量标準制定依據
5 批檢驗報告
6 自身穩定性研究
7 相容性和安全性研究
7.1 相容性研究
7.2 安全性研究
附表1高風險藥包材使用情況與登記資料表
附表2非高風險藥包材使用情況與登記資料表
附表3實行關聯審評審批的藥包材及風險分類
二、登記資料正文及撰寫要求
1 登記人基本信息
1.1 名稱、注冊地址、生産廠、生産地址
提供登記人的名稱、注冊地址。
提供生産廠的名稱、生産地址(如有多個生産場地,都應提交)。
生産地址應精確至生産車間。
1.2證明性文件
境内藥包材登記人需提交以下證明文件:
登記人營業執照複印件,營業執照應包含此次登記産品。對登記人委托第三方進行生産的,應同時提交委托書等相關文件、生産者相關信息及營業執照。
境外藥包材登記人應授權中國代表機構提交以下證明文件(參照進口藥品注冊有關規定):
(1)登記人合法生産資格證明文件、公證文件及其中文譯文。對登記人委托第三方進行生産的,應同時提交生産者相關信息及證明文件(如适用)。
(2)登記人如委托中國境内代理機構登記,需提交授權文書、公證文件及其中文譯文。中國境内代理機構需提交其工商執照或者注冊産品生産廠商常駐中國境内辦事機構的《外國企業常駐中國代表機構登記證》。
(3)産品在境外的生産、銷售、應用情況綜述及在中國申請需特别說明的理由。
1.3 研究資料保存地址
提供研究資料保存地址,應精確至門牌号。如研究資料有多個保存地址的,都應提交。
2 藥包材基本信息
採用相同的生産工藝和材料、具有相同功能的産品可以作爲同一藥包材登記,藥包材企業可在同一登記号下按不同的型号和規格進行登記。
2.1 藥包材名稱
提供藥包材的中英文通用名稱、化學名稱(如适用)、曾用名,對於尚無法確定通用名稱的,需提供拟定名稱。藥包材名稱應與品種質量标準中的名稱一緻,也可參考主管部門制定的命名原則進行命名。應當參照已批準的藥包材名稱或國家标準命名原則對産品進行命名。
2.2 包裝系統/組件
藥包材可以是包裝系統,也可以是包裝組件,組件需說明适用的包裝系統。
包裝系統/組件需分别提供每一個單獨組件/材料的相關信息,包括構成系統的組件産品名稱、來源、生産地址等相關信息及質量标準、檢驗報告等。如果有多個來源,需分别列出或提供組件的登記号。
說明:請按照附件填寫包裝系統各包裝組件的名稱。如:經口鼻吸入制劑應填寫容器(如罐、筒)、閥門等配件。
對於某些制劑,如需在直接接觸藥品的藥包材外增加功能性次級包裝材料(如高阻隔性外袋),或者需包裝初級以及次級包裝材料後進行滅菌處理的制劑,需将初級以及次級包裝材料作爲包裝系統,一並進行填寫,例如某些採用初級及次級塑料包裝材料的注射制劑,對於所用的幹燥劑、吸氧劑、指示劑等,也應填寫,如影響制劑質量的,需訂入包材的質量标準中。制劑生産過程中不參與滅菌處理,僅爲防塵用的外袋,可不作爲功能性次級包裝材料。
2.3 配方
應分别填寫藥包材中各個組件的配方信息,包括組分名稱、來源、質量标準及檢驗報告、用量配比和預期用途、化學品安全說明書(MSDS)。有登記号的組件也可提供登記号。配方信息應覆蓋藥包材所涉及的所有組成部分及用量依據,如添加劑在境内外藥典、國标等法規标準收載的用量範圍。
2.3.1名稱:包括原輔料及添加劑(著色劑、防腐劑、增塑劑、遮光劑及油墨等)的化學名(IUPAC名和/或CAS名)、中文譯名和商品名等。
說明:原輔料名稱中應同時注明該原料的使用等級(如有,需提供),聚合物和金屬材料應注明牌号。
2.3.2來源:提供原輔料的生産商,分析原輔料的作用。
2.3.3相對分子量、分子式、化學結構:未應用於相同給藥途徑的系統或組件中的新物質需提供化學結構的確認依據(如核磁共振譜圖、元素分析、質譜、紅外譜圖等)及其解析結果。
2.3.4理化性質:包括各組分的理化性質,如顔色、氣味、狀态、溶解度、分子量、聚合度等(如适用可提供)。
2.3.5用量配比和預期用途:對原輔料的用量/用量範圍/比例進行說明,並對其在材料生産、加工及使用過程中所起到的作用分别進行描述。
2.3.6化學品安全說明書(MSDS):應提供原輔料生産廠家提供的或從公開途徑獲得的所使用各種物質的化學品安全說明書。提供配方彙總表,示例如下:
配方彙總表
組件一:膠塞 | |||||
a 主要原料 | 來源 | 标準 | 用量 | 用途 | 生産商 |
b 輔料 | |||||
注:來源是指制備材料的來源,如:天然(動植物)或人工合成等。
2.4 基本特性
2.4.1基本信息
根據具體藥包材種類,分别提供藥包材以及各組件的基本特性信息。
例如:對於吸入制劑,應填寫整體藥包材的相關物化性質,如外觀、尺寸、形狀、顔色、組成、規格、用途等,還應填寫閥門等組件的相關物化性質(具體可參考藥包材的相關技術指導原則)。
2.4.2保護性和功能性
如果登記的是包裝組件,保護性和功能性可能需要由包裝系統的組合單位進行相應研究。
保護性:藥包材應保證對藥品制劑在生産、運輸、儲存及使用過程中的保護性能,包括光線、溫度、濕度以及在受力條件下對材料及容器保護性能的影響進行相關研究(或提供長期上市使用的證明或相關文獻資料)。
需根據藥包材的用途,提供相應的保護性和功能性研究資料,以及方法學驗證資料(如适用)。如:避光防護、防止溶劑流失/滲漏、保護滅菌産品或有微生物限度要求的産品免受細菌污染、防止産品接觸水汽、防止産品接觸反應性氣體等。說明藥包材質量标準中是否有相應的質控項目。例如:透光率,氧氣、水分、氮氣、二氧化碳透過率等密閉性能的驗證數據等。對於需滅菌處理的無菌制劑用包裝,必須提供滅菌工藝适應性的驗證資料,目前常用的滅菌工藝包括環氧乙烷滅菌、濕熱滅菌、輻射滅菌等,需考察滅菌工藝對材料的影響(是否适合滅菌過程),環氧乙烷滅菌還需考察環氧乙烷及其相關物質的殘留情況。終端滅菌制劑包裝需提供溫度适應性研究資料,並在質量标準中列出可耐受的滅菌條件等信息。如适用,無菌制劑用包裝還需要進行滅菌效果的驗證,並對包裝材料的微生物學性質進行研究,從而確定無菌包裝的儲存期。
功能性:藥包材功能性是指包裝系統按照預期設計發揮作用的能力,如滿足特殊人群(兒童、老年人、盲人等)用藥、提高患者用藥依從性以及附帶給藥裝置的性能。
需根據藥包材的用途,提供相應的功能性研究資料,以及方法學驗證資料。如果採用國家标準/行業标準試驗方法,則不需提供方法學驗證資料。
對於具有特定功能的包裝,如控制藥物釋放的噴霧劑定量給藥裝置、帶高阻隔性外袋的塑料藥包材等,需提供針對特定功能進行的相關驗證資料,以滿足特定的功能性要求。對於提高用藥依從性,降低錯誤用藥的包裝形式,如兒童安全蓋、粉液雙室袋、盲文印刷、老年人易開啓等,還應提供操作可行性實驗分析以及一定人群範圍的應用數據分析。
2.5 境内外批準及使用信息
2.5.1境外批準上市的相關證明性文件
對於進口藥包材,提供境外藥品監督管理部門的相關證明性文件,如DMF備案文件(說明狀态)、批準時間和/或其他證明性文件,並簡述在制劑中的使用情況。
2.5.2生産、銷售、應用情況綜述
填寫本企業所生産藥包材在境内上市(包括進口)的制劑中是否已經應用,以及所應用的劑型、産品。
2.6 國家标準以及國内外藥典收載情況
提供該藥包材及各組件被國家标準及國内外藥典以及相關國際标準收載的信息。
3 生産信息
3.1 生産工藝和過程控制
提供生産廠區及潔淨室(區)平面圖及潔淨區的檢測報告。
(1)工藝流程圖:按工藝步驟提供工藝流程圖,标明工藝參數、關鍵步驟等。若使用溶劑請列出所用溶劑種類。
(2)工藝描述:根據工藝流程來描述工藝操作,以商業批爲代表,列明主要工藝步驟、添加劑、粘合劑、生産條件(溫度、壓力、時間等)和操作程序等,如産品涉及印刷,需說明印刷工藝及採用的印刷介質等相關信息,說明生産工藝的選擇依據。
滅菌的藥包材可增加包材使用前清洗狀态及烘幹、滅菌要求等。
(3)說明商業化生産的分批原則、批量範圍和依據。
(4)設備:提供主要和特殊的生産、檢驗設備的型号或技術參數。
生産、檢驗設備資料可以按照下述表格形式提交:
藥包材生産設備一覽表
序号 | 設備名稱 | 型号 | 數量 | 生産廠 |
1 | ||||
2 | ||||
… |
藥包材檢驗設備一覽表
序号 | 設備名稱 | 型号 | 數量 | 生産廠 |
1 | ||||
2 | ||||
… |
3.2 物料控制
按照工藝流程圖中的工序,以表格的形式列明生産中用到的所有物料和添加劑,如油墨和粘合劑等並說明所使用的步驟,示例如下:
物料控制信息
物料名稱 | 來源注 | 質量标準 | 生産商 | 使用步驟 |
注:如動物來源、植物來源、化學合成等。
提供以上物料的質量控制信息,明確引用标準,或提供内控标準(包括項目、檢測方法和限度)並提供必要的方法學驗證資料。
3.3 關鍵步驟和半成品/中間體的控制
列出所有關鍵步驟,提供關鍵過程控制及參數,提供可確定關鍵步驟合理性以及工藝參數控制範圍合理性的研究資料。需說明各生産步驟是否爲連續生産。
如有半成品/中間體,列出半成品/中間體的質量控制标準,包括項目、方法和限度,並提供必要的方法學驗證資料。
3.4 工藝驗證和評價
對産品質量有重大影響的工藝,應提供驗證方案、驗證報告、批生産記錄等資料,或提供足夠信息以證明生産工藝能穩定生産出符合質量要求的包材。
4 質量控制
4.1 質量标準
提供藥包材的标準:
已有國家标準的登記産品,可使用國家标準作爲登記産品的質量标準,如果與國家标準或藥典标準不一緻,需結合産品的性質,論證企業标準確定的合理性。登記産品的材料、用途、生産工藝(适用時)、組合件配合方式(适用時)的要求應與質量标準的規定相符。
關於企業标準的要求:
質量标準應當符合現行版中國藥典和國家标準的技術要求和格式,並使用其術語和計量單位。尚未收入國家标準的登記産品,登記企業應根據登記産品的材質、用途、性能等特點,設立相關檢驗項目、檢驗方法和技術要求,自行拟定産品注冊标準,並進行方法學驗證;
提供标準編制和起草說明,提供項目、方法、指标設立的依據等内容。同一個包裝系統/組件用於不同的制劑或不同的制劑企業,檢測項目和指标可能不同。安全性指标應不得低於國家标準同類産品的要求;
根據藥包材産品種類及其适用劑型的不同,在質量标準中需包含材料、容器的阻隔性能和密閉性能等相應的保護性檢測項目:如避光、防潮、隔絕氣體(氧氣、水分、氮氣、二氧化碳透過率等)、密閉、防止微生物污染等保護性檢測項目。可使用藥典等方法進行透光性,防潮性,微生物限度和無菌測試。必要時,除藥典等标準裏列出的這些測試以外,可以增加有關性能測試(如氣體傳導,溶劑滲漏,容器完整性);
質量标準中需包含藥包材安全性、保護性、功能性與生産過程相關的檢測項目;
提供産品的結構示意圖(包括尺寸信息)和實樣圖片。
4.2 分析方法的驗證
提供質量标準中相關項目的方法學驗證資料。某些無需進行驗證的檢查項,如酸堿度滴定,水分測定等,無需提供;對於採用國家藥包材标準的分析方法無需提供分析方法的驗證;對於採用相關國際标準或國外藥典收載的方法,可視情況開展方法學確認。
4.3 質量标準制定依據
企業标準需要說明各項目設定的考慮,總結分析各檢查方法選擇以及限度確定的依據。質量标準起草說明應當包括标準中控制項目的選定、方法選擇的依據等。
5 批檢驗報告
逐批檢驗項目需提供不少於三批樣品的檢驗報告,YBB中*号檢驗項目可以檢一批。如果委托有資質單位進行檢驗的項目需予以說明。委托檢驗的受托方需具備相關資質。
6 自身穩定性研究
提供藥包材自身的穩定性研究資料,描述針對所選用包材進行的支持性研究。
藥包材自身穩定性重點考察包裝系統或包裝組件在規定的溫度及濕度環境下随時間變化的規律,以確認藥包材産品在規定的貯存條件下的穩定期限。
說明穩定性研究的樣品情況(包括批号、批量等信息)、考察條件、考察指标和考察結果,對變化趨勢進行分析,並提出貯存條件和使用期限。以表格形式提供穩定性研究的具體結果,並将穩定性研究中的相關圖譜作爲附件。
藥包材穩定性研究可參照相關技術指導原則進行,如加速條件下的老化研究,也可提供藥包材在穩定期内的長期試驗數據。穩定性評價的樣品應具有代表性,通常應採用穩定規模生産的樣品。樣品的質量标準應與規模生産所使用的質量标準一緻。
藥包材自身穩定性研究一般适用於藥用塑料和橡膠等高分子材料。
7 相容性和安全性研究
7.1 相容性研究
用於吸入制劑、注射劑、眼用制劑的藥包材,登記人應根據配方提供提取試驗信息,包括定量或定性地獲取材料中揮發性或不揮發性提取物的提取特性(提取物庫)以及相應的譜圖。如有可能,可同時提供潛在的浸出物提示信息,供制劑生産企業進行制劑與藥包材的相容性試驗使用。
提取試驗可參照國家發布的相關技術指導原則或國内外藥典收載的相關标準進行。提取試驗的方法和溶劑的選取應根據提取目的和包裝組件的性質決定。理想情況下提取溶劑應與制劑對提取物質具有相同的特性以獲得同樣的定量提取特征。
7.2 安全性研究
下列産品需要進行安全性研究:
7.2.1新材料,新結構,新用途的藥包材:應提供産品及所用原材料相關的安全性(生物學和毒理學)研究資料,具體産品安全性研究資料可參考相應技術要求進行。
7.2.2用於吸入制劑、注射劑和眼用制劑的藥包材:無明確證據應用於此類包裝的材料和添加劑,需提供相應的安全性資料。爲證明相容性,對有可能發生藥品與包裝材料發生相互作用的情況,應提交可提取物的毒理學研究,必要時提供可提取物的生物學安全性評價資料;應提交已知可提取物的結構(包括結構已知且毒理學數據明確的可提取物,以及結構已知但毒理學數據不明確的可提取物)。
三、登記資料說明
1 藥包材登記資料是制劑注冊資料中的一部分,與制劑的登記資料組合後,應能夠證明該産品可以滿足預期包裝藥品的要求。
2 登記資料依據藥包材風險程度和藥包材使用情況進行分類,確定至少需要提交的登記資料,具體内容見附表1和附表2。
3 藥包材産品及所用原材料、添加劑相關的安全性研究資料,可包括以下資料:國内外藥包材标準或藥典中的生物學測試;國内外毒理學文獻資料;材料的生物學安全性測試等。
4 對於非高風險制劑使用的藥包材,暫不要求提供3.4工藝驗證和評價及7相容性和安全性研究資料。採用無菌工藝的外用制劑,以及所有的液體制劑使用的藥包材應視情況開展相應的研究。
5 如果申請的藥包材涉及多個組件組成包裝系統,除包裝系統要填報完整的登記資料外,每個組件需分别提供資料2.2~7。例如大容量注射劑的輸液袋包裝,需分别填寫多層共擠輸液袋、塑料組合蓋、阻隔外袋等信息。如果僅登記包裝組件,如藥用膠塞,可僅填寫膠塞的相關信息。
6 對於分類未涵蓋的藥包材,請選擇“其他”,其登記資料要求根據風險程度和使用曆史提交相關的登記資料。
附表1
高風險藥包材使用情況與登記資料表
資料項目 | 資料内容 | 1* | 2* | 4.1* | 4.2* |
1登記人基本信息 | 1.1 名稱、注冊地址、生産地址 | + | + | + | + |
1.2 證明性文件 | + | + | + | + | |
1.3 研究資料保存地址 | + | + | + | + | |
2藥包材基本信息 | 2.1 藥包材名稱 | + | + | + | + |
2.2 包裝系統/組件 | + | + | + | + | |
2.3 配方 | + | + | + | + | |
2.4 基本特性 | + | + | + | + | |
2.5 境内外批準及使用信息 | - | + | + | + | |
2.6 國家标準以及國内外藥典收 載情況 | + | + | + | + | |
3生産信息 | 3.1 生産工藝和過程控制 | + | + | + | + |
3.2 物料控制 | + | + | + | + | |
3.3 關鍵步驟和半成品/中間體的控制 | + | ± | - | - | |
3.4 工藝驗證和評價 | + | ± | - | - | |
4質量控制 | 4.1 質量标準 | + | + | + | + |
4.2 分析方法的驗證 | + | + | + | - | |
4.3 質量标準制定依據 | + | + | - | - | |
5批檢驗報告 | + | + | + | + | |
6自身穩定性研究 | + | + | + | - | |
7相容性和安全性研究 | 7.1 相容性研究 | + | + | + | - |
7.2 安全性研究 | + | - | + | - | |
注:+ 需提供相關資料的項目
- 無需提供相關資料的項目
± 根據需要提供相關資料的項目
附表2
非高風險藥包材使用情況與登記資料表
資料項目 | 資料内容 | 1* | 2* | 3* | 4.1* | 4.2* |
1登記人基本信息 | 1.1名稱、注冊地址、生産地址 | + | + | + | + | + |
1.2證明性文件 | + | + | + | + | + | |
1.3研究資料保存地址 | + | + | + | + | + | |
2藥包材基本信息 | 2.1藥包材名稱 | + | + | + | + | + |
2.2包裝系統/組件 | + | + | + | + | + | |
2.3配方 | + | + | + | + | + | |
2.4基本特性 | + | + | + | + | + | |
2.5境内外批準及使用信息 | - | + | - | + | + | |
2.6國家标準以及國内外藥典收載情況 | + | + | + | - | - | |
3生産信息 | 3.1生産工藝和過程控制 | + | + | + | + | + |
3.2物料控制 | + | + | + | + | + | |
3.3關鍵步驟和半成品/中間體的控制 | + | - | - | - | - | |
3.4工藝驗證和評價 | + | - | - | - | - | |
4質量控制 | 4.1質量标準 | + | + | + | + | + |
4.2分析方法的驗證 | + | + | + | + | - | |
4.3質量标準制定依據 | + | + | + | - | - | |
5批檢驗報告 | + | + | + | + | + | |
6自身穩定性研究 | + | + | + | + | - | |
7相容性和安全性研究 | 7.1相容性研究 | - | - | - | - | - |
7.2安全性研究 | + | ± | - | - | - | |
注:+ 需提供相關資料的項目
- 無需提供相關資料的項目
± 根據需要提供相關資料的項目
備注:*
1未在境内外上市藥品中使用過的藥包材(如新材料、新結構);
2已在境内外上市藥品中使用過,但改變藥品給藥途徑且風險提高的藥包材;
3未在境内外上市藥品中使用過,但是可證明在食品包裝中使用過的與食品直接接觸的藥包材(僅限用於口服制劑);
4已在相同給藥途徑的上市藥品中使用過的藥包材
4.1無注冊證的藥包材
4.2有注冊證的藥包材
附表3
實行關聯審評的藥包材及風險分類
制劑類别 | 劑型 | 包裝系統 | 包裝組件 |
經口鼻吸入制劑 | 氣霧劑、噴霧劑、粉霧劑 | 吸入制劑密閉系統 | 罐(筒)、閥門 |
注射制劑 | 小容量注射劑 | 預灌封注射劑密閉系統 | 針筒(塑料、玻璃)、注射鋼針(或者魯爾錐頭)、活塞 |
筆式注射器密閉系統 | 卡式玻璃瓶+玻璃珠、活塞、墊片+鋁蓋 | ||
抗生素玻璃瓶密閉系統 | 玻璃瓶、膠塞、鋁蓋(或者鋁塑組合蓋) | ||
玻璃安瓿 塑料安瓿 | |||
大容量注射劑 | 玻璃瓶密閉系統 | 玻璃瓶、膠塞、鋁蓋(鋁塑組合蓋) | |
軟袋密閉系統 | 多層共擠輸液膜、塑料組合蓋、膠塞、接口 | ||
塑料瓶密閉系統 | 塑料瓶、塑料組合蓋 | ||
沖洗液、腹膜透析液、腸内營養液等 | 軟袋密閉系統 | 輸液膜、塑料組合蓋或者其他輸注配件 | |
眼用制劑 | 眼用液體制劑 | 塑料瓶密閉系統 | |
其他眼用制劑,如眼膏劑等 | 眼膏劑管系統 | 軟膏管、蓋、墊片 | |
透皮制劑 | 貼劑 | 透皮制劑包裝系統 | 基材、格拉辛紙+複合膜 |
口服制劑 | 口服固體制劑 | 塑料瓶系統、 玻璃瓶系統 | |
複合膜袋 | 複合膜 | ||
中藥球殼 | |||
泡罩包裝系統 | 泡罩材料、易穿刺膜 | ||
口服液體制劑 | 塑料瓶系統、 玻璃瓶系統 | 瓶身、瓶蓋、墊片 | |
外用制劑 | 氣霧劑、噴霧劑、粉霧劑 | 外用制劑密閉系統 | 罐(筒)、閥門 |
軟膏劑、糊劑、乳膏劑、凝膠劑、洗劑、乳劑、溶液劑、搽劑、塗劑、塗膜劑、酊劑 | 外用制劑包裝系統 | ||
藥用幹燥劑 | |||
其他 | |||
注:
1.高風險藥包材一般包括:用於吸入制劑、注射劑、眼用制劑的藥包材;國家藥品監督管理局根據監測數據特别要求監管的藥包材;新材料、新結構、新用途的藥包材參照上述要求執行。
2.鼓勵按照包裝系統進行登記,如因爲技術原因不能按照完成的包裝系統登記,也可按照包裝組件進行登記。
附件3
可選擇不按照146号公告要求
進行登記的部分矯味劑、香精、色素、pH
調節劑等藥用輔料
(2019年版)
藥品制劑所用的部分矯味劑、香精、色素、pH調節劑等藥用輔料可不按照146号公告要求進行登記,具體如下:
1.矯味劑(甜味劑):如蔗糖、單糖漿、甘露醇、山梨醇、糖精鈉、阿司帕坦、三氯蔗糖、甜菊糖苷、葡萄糖、木糖醇、麥芽糖醇等。該類品種僅限於在制劑中作爲矯味劑(甜味劑)使用。
2.香精、香料:如桔子香精、香蕉香精、香蘭素等。執行食品标準的,應符合現行版GB 2760《食品安全國家标準 食品添加劑使用标準》、GB 30616《食品安全國家标準 食品用香精》及GB 29938《食品安全國家标準 食品用香料通則》等相關要求。
3.色素(著色劑):如氧化鐵、植物炭黑、胭脂蟲紅等。執行食品标準的,應符合現行版GB 2760《食品安全國家标準 食品添加劑使用标準》等相關要求。
4.pH調節劑(包括注射劑中使用的pH調節劑):如蘋果酸、富馬酸、醋酸、醋酸鈉、枸橼酸(鈉、鉀鹽)、酒石酸、氫氧化鈉、濃氨溶液、鹽酸、硫酸、磷酸、乳酸、磷酸二氫鉀、磷酸氫二鉀、磷酸氫二鈉、磷酸二氫鈉等。
5.僅作爲輔料使用、制備工藝簡單、理化性質穩定的無機鹽類(包括注射劑中使用的無機鹽類):如碳酸鈣、碳酸鈉、氯化鉀、氯化鈣、氯化鎂、磷酸鈣、磷酸氫鈣、硫酸鈣、碳酸氫鈉等。
6.口服制劑印字使用的無苯油墨。
上述藥用輔料,現行版《中國藥典》已收載的,應符合現行版《中國藥典》要求;現行版《中國藥典》未收載的,應符合國家食品标準或現行版美國藥典/國家處方集、歐洲藥典、日本藥典、英國藥典标準要求;其他輔料,應符合藥用要求。
附件4
藥用原輔料、藥包材年度報告基本要求
一、年度報告應在登記平台公示後第12個月通過申請人之窗提交。
二、年度報告中應包括12個月内産品變更彙總,如無任何變更應有相關聲明。
三、年度報告中應有相關制劑産品信息,如企業名稱、藥品名稱等。